

24 de Janeiro
de 2022 Robert Bibeau

"CTCCTCGGGGGGCACGTAG" é a arma
fumegante de Covid-19 que fará luz sobre esta pandemia.

O genoma SARS-CoV-2 contém uma
sequência de 19 nucleótidos #CTCCTCGGCGGGCACGTAG
probabilidade de sequência que ocorre por
acaso: menos de 1 em um bilião
não há nenhum vírus conhecido, incluindo
esta sequência antes de SARS-Cov-2
Moderna tem patentes anteriores à pandemia
que incluem esta sequência
— bitbutter (@mormo_music) 14 de Janeiro de 2022

O link para aceder a esta página foi removido
De acordo com uma extensa pesquisa conduzida pelo Dr. Ah Kahn Syed, a Moderna
patenteou a sequência única encontrada no coronavírus dois anos antes da
pandemia começar, reforçando a teoria de que Covid-19 é um vírus feito pelo
homem.
O Dr. Syed escreve: há muito tempo que queria escrever este
artigo. Bem, pelo menos desde que Prashant Pradhan (um maravilhoso, honesto e
corajoso cientista de genómica) levantou a possibilidade, em Fevereiro de 2020,
de que o vírus SARS-Cov2 foi feito pelo homem. E vimos muitas peças confirmando
que o vírus foi fabricado em laboratório, um dos melhores aqui em zenodo e com o seu próprio vídeo aqui. No momento da escrita, estas ligações
ainda estão em vigor, o que, ao fim de 12 meses, é uma coisa boa para qualquer
artigo que se atreva a questionar os mexericos propagados pela nossa
querida "imprensa
livre [patrocinada pela indústria farmacêutica]".
De qualquer forma,
BLAST é o repositório NCBI/NIH (também conhecido como governo dos EUA) para
sequências genómicas e proteómicas, entre outras coisas. É aqui que todos os
geneticistas do mundo depositam as suas sequências quando fazem uma descoberta.
A sua principal função é permitir a comparação de sequências genéticas e a
descoberta de sequências que correspondem a uma sequência que pode ter
encontrado na sua experiência. O que é uma sequência genética? É simples. É uma
linha de código, composta por qualquer combinação de 4 letras numa sequência.
Lembra-se do filme GATTACA? Se ainda não o viu, devia, porque este
é outro filme distópico que agora está demasiado perto de casa.
O título do filme baseia-se nas 4 bases de nucleótidos (G, A, T, C) que
compõem o código genético do ADN de cada ser humano. Existem cerca de 3 mil
milhões deles em cada célula, resultando num código único – o que faz de si um
indivíduo único! O código combina de tal forma que G-C e A-T combinam sempre
para formar a dupla hélice que se vê na imagem, por exemplo GATTACA seria
emparelhado com CTAATGT (o complemento). O código é lido num sentido
específico, pelo que o GATTACA numa vertente seria o TGTAATC na outra (o
complemento inverso). Uma das vantagens do BLAST é que não se importa com a
versão que lhe dá, dir-lhe-á sempre o gene certo.
Outra coisa a notar
neste momento são as probabilidades. Você fez isso na escola atirando uma moeda
(onde o código é H para coroa ou T para cara). Qual seria a probabilidade de
HHHHH (1 em 2^4 = 1/16). O mesmo se aplica ao TTTT. O mesmo se aplica ao THTH,
ou a qualquer outra sequência específica de impressões de coroa ou cara.
Experimente-o se não acredita em mim (preveja primeiro a sequência e depois
veja quantas vezes tem de a executar). O código genético é essencialmente
uma "moeda
de quatro lados". Assim, para qualquer execução de uma sequência
específica (por exemplo, GATC), a probabilidade de obter esta sequência EXACTA
é de 1 em 4^4, ou para qualquer número n de nucleótidos (nt ou bases), a
probabilidade é de 1 em 4^n (isto é simplificado porque em algumas situações, a
probabilidade de que a próxima base é X depende das bases circundantes).
Blast tem duas secções - nucleótido (BLASTn) e proteína (BLASTp). BLASTp
trata as sequências de aminoácidos da mesma forma que as sequências de
nucleótidos. Mas há uma grande diferença porque existem 20 aminoácidos (em vez
de 4 nucleótidos) e, portanto, mesmo sequências curtas (por exemplo, QTNS =
Glu-Thr-Asn-Ser) teriam uma probabilidade de cerca de 1 em 20^4 (simplificado),
ou 1 em 160.000. A probabilidade de uma sequência específica de 5 aminoácidos
aparecer por acaso na mesma base é de 1 em 3,2 milhões!

Vamos premir o botão Protein BLAST e vamos partir... e aqui está o ecrã que vai ter, que eu o vou obrigar a navegar.

Em [1], é necessário introduzir a sequência de aminoácidos em que está interessado (BLAST adiciona automaticamente ">produto proteico sem-nome "). Felizmente, não precisa de olhar muito, pois vamos focar-nos em apenas 4 sequências no genoma viral/proteome da SARS-CoV-2 e estas estão em destaque no maravilhoso artigo de Prashant Pradhan "Semelhança estranha de inserções únicas na proteína spike 2019-nCoV para HIV-1 gp120 e Gag" publicado em 31 de Janeiro, 2020 poucos dias depois da sequência do genoma ter sido publicada.
A parte que precisa está na tabela 1 que publico aqui e verá que apresentei
a sequência de 6 aminoácidos TNGTKR na caixa [1] marcada a vermelho no ecrã
BLASTp.

Para o passo [2] do ecrã de entrada BLASTp, é necessário adicionar alguns filtros. O primeiro filtro é restringir a pesquisa a "viridae (vírus)" (ou simplesmente pode introduzir 10239, que é o ID taxonómico). A razão para isto é que há biliões de espécies no planeta e o BLASTp vai procurá-las todas, mas você só quer saber de que vírus este padrão vem. Não é realmente interessante saber se o padrão é encontrado numa lula, embora seja possível que uma lula também tenha participado na acção com o famoso morcego e o pangolim, de que pessoas como Peter Daszak e Dominic Dwyer falam, tornando-o uma menagerie zoonótica, mas vamos quedar-nos na realidade.
A segunda condição é excluir todas as referências à SARS-CoV-2 que agora se
acumularam na base de dados, porque todas elas aparecerão (milhares) e não
estamos interessados.
Depois de introduzir estes dados, prima o botão BLAST e o que obtém?
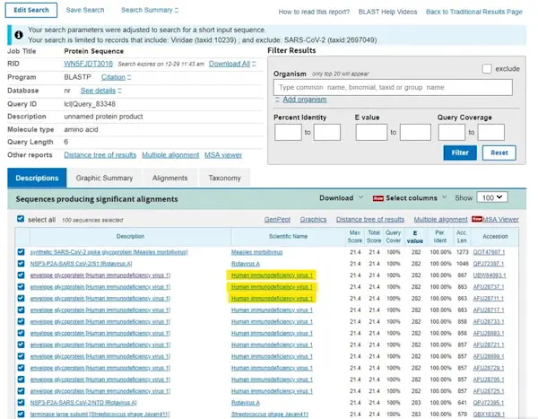
Receberá uma lista de candidatos que têm uma homologia próxima desta sequência.
Como esta é uma sequência muito curta, a homologia (semelhança) deve ser 100%.
O topo da página é um resumo do que pediu e o resto da página é uma lista de
sequências correspondentes. O que você verá imediatamente é que no topo da
lista estão dois vírus sintéticos que são uma quimera de SARS-Cov-2 e outro
vírus, que apareceu nos últimos dois anos graças a laboratórios que fabricam
outros vírus (porque não temos o suficiente). O próximo da lista é um monte de
referências ao HIV-1.
Pode clicar em qualquer um deles e será levado para o ecrã de alinhamento
onde são apresentados os alinhamentos entre o sujeito (o seu TNGTKR) e a
consulta (todos os vírus), e verá à medida que vai descendo a página que os
alinhamentos só se mantêm para o HIV-1 até começar a obter proteínas sintéticas
e hipotéticas, até o próximo vírus real na lista que é HIV-2....
Está bem, mas um
resultado como este pode ser uma coincidência. Na lista, verá o "valor E" que é um
indicador da probabilidade de encontrar fósforos como este e deve estar o mais
perto de zero possível. Aqui é 282, o que só reflete a probabilidade de
encontrar correspondências numa sequência curta.
Então este é o problema. Ou estas sequências corresponderão, por acaso, a
um monte de outros vírus (porque o valor E é elevado e, portanto, temos de
esperar muitas correspondências), ou são sequências realmente incomuns que
especificamente, preferencialmente ou apenas corresponderam ao HIV-1. Como
abordar esta questão? Bem, vamos passar para a próxima sequência : HKNNKS, que
é outra sequência curta. Para colocar o ecrã em ordem, podemos definir um
filtro para sequências curtas para garantir que 100% da sequência corresponde
(direita superior). Agora, lembre-se que se o jogo com o HIV-1 foi aleatório,
não devemos realmente ver qualquer correspondência nesta lista, pois teria que
ser eliminado por todas as outras centenas de vírus que devem corresponder
preferencialmente. Oops...

Observe as outras correspondências – Bat RaTG13 que só apareceu nesta base
de dados depois de as pessoas começarem a questionar a origem do coronavírus, e
que é provavelmente uma sequência sintética, e os mesmos vírus sintéticos que
apareceram após o surto. Então o HIV-1 é o único par para estas duas
sequências.
Vamos passar à sequência 3. É uma sequência mais longa. RSYLTPGDSSG.
Pergunto-me qual vírus (ou que conjunto de vírus aleatórios, porque é uma
sequência aleatória, lembre-se) que corresponde ..... Oh, olhe...

Nesta pesquisa, limitei a cobertura da consulta a 100% para eliminar o ruído e proteínas hipotéticas. Resta-nos apenas os vírus sintéticos do pós-covid e do RaTG13 (também pós-covid). O único vírus que resta nesta lista é, adivinhou, o HIV-1. Quais são as hipóteses de o HIV-1 aparecer nas 3 buscas?
E para completar a
ronda quádrupla, precisamos da última sequência de inserção identificada no
artigo de Pradhan, nomeadamente QTNS-PRRA. Esta é uma sequência muito
interessante à qual voltaremos mais tarde, porque é o local de clivagem do
furin. É interessante porque os coronavírus beta como este não têm um local
de clivagem
da furina, é o único. Certamente este site não pode vir do HIV-1? Bem, não vem da
proteína GP120 como as outras três sequências, é completamente diferente e está
noutro lugar no vírus, que eu vou mostrar-lhe em breve, mas por agora, vamos
executar BLASTp.
Desta vez é um pouco mais complicado porque muitas proteínas hipotéticas e
sintéticas foram adicionadas desde a publicação do SARS-CoV-2 (eu deveria ter
escrito este artigo no ano passado). Mas o HIV-1 está a voltar à lista, e desta
vez só vou mostrar os alinhamentos entre a proteína de brincadeira e o
coronavírus – neste caso, há uma eliminação na proteína HIV-1.

Portanto, temos 4
correspondências com sequências de HIV sem qualquer outro vírus* que apareça nas 4 listas de
correspondências (*excepto vírus sintéticos criados após o evento). Quais são as
hipóteses disto - perto de zero.
Mas olhe para aquilo.
Não eram apenas sequências aleatórias de HIV. No seu trabalho, Pradhan foi mais
longe e recriou a estrutura do vírus com a localização das quatro inserções. E
agora estas inserções
"aleatórias",todas do HIV, estão todas em sites de ligação
coronavírus. Quais são as hipóteses?

Pode não estar convencido. Apesar de o único vírus aparecer nas listas de
correspondência para as quatro inserções, entre as centenas de milhares de
vírus existentes, acontece ser o HIV-1. E o HIV-1 não deve ter hipóteses reais
de formar vírus recombinantes com coronavírus de morcegos na natureza, e
nenhuma chance real de formar 4 recombinações diferentes que estão apenas em
locais de ligação para o vírus. Mas se isso não o convencer, há uma
característica especial do inserível 4 que precisamos olhar.
Agora voltamos aos
nucleótidos, o G-A-C-T que compõem a sequência de aminoácidos de que falámos
até agora. A sequência de referência original do genoma coronavírus é
identificação na base de dados NC_045512.1 e pode ser consultada no seguinte
endereço: https://www.ncbi.nlm.nih.gov/nuccore/NC_045512.1
Pode reproduzir com a
sequência do genoma utilizando o BLASTn (para nucleótido) indo a esta página e
selecionando "Run
BLAST" na coluna direita, que o levará a uma página BLAST semelhante à das
proteínas acima.

Da mesma forma,
introduza NC_045512.1 (ou atualização NC_045512.2) na primeira caixa, escolha
as opções indicadas na parte marcada [2], seleccione o megablast na parte 3 e
clique em ir, e receberá uma lista de genomas SAR-CoV-2 que correspondem
(obviamente). Não vou mostrar este ecrã porque não é importante aqui, mas aqui
está o ecrã que obtém se vir duas sequências intimamente emparelhadas e clicar
em "recurso
CDS" para sobrepor a sequência de aminoácidos. Vais acabar com páginas
assim:

Nesta secção em
particular, pode ver-se que se trata de uma sequência da "proteína spike" (glicoproteína
superficial) e que os nucleótidos estão rotulados 23548... 23771
(aproximadamente 30.000 nucleótidos ou bases, ou seja, G-C-A-T). [NB: na
verdade, é RNA e deve haver um U em vez de cada T, mas BLAST compensa automaticamente
para simplificar]. O menor número é o número de aminoácidos na sequência
proteica, por isso, para cada 3 nucleótidos, o número aumenta por um
aminoácido. O ponto forte é o aminoácido 677 (Q) a 686 (S), que dá 677→686 =
QTNSPRRARS.
Agora, é realmente
interessante porque não só vimos que a secção QTNS é derivada do HIV, mas há
algo muito especial sobre o PRRAR adjacente porque é um local de clivagem de furina e, como vimos,
estes não existem neste tipo de vírus tipo SARS. Esta é uma inserção no genoma
viral, mas ninguém realmente sabe como chegou lá (assim como as sequências de
HIV). Para saber de onde vem, precisamos olhar para além da sequência de
aminoácidos e voltar para a sequência do genoma.
A sequência genómica
que pode ver para esta sequência de aminoácidos é: CAGACTAATTCCCCTCGGGGGGGGGGGGGGGGGGCGTAGT,
ou 30 nucleótidos codificando 10 aminoácidos. Para que esta sequência fosse o
resultado do acaso, seria preciso um número infinitamente pequeno. Deve,
portanto, ter aparecido algures (ou seja, de outro vírus) ou parte desta sequência
deve ser sintética. Vamos, portanto, analisá-lo pela BLAST(n), desta vez
excluindo "construcções
sintéticas" da nossa pesquisa (porque estamos à procura de vírus reais, não vírus
sintéticos). O que temos aqui?

Agora que está
habituado a estes ecrãs, podemos ver que as únicas sequências virais presentes
aqui são sintéticas, e se clicar em cada uma delas, encontrará a data de
registo depois de Fevereiro de 2020. Por outras palavras, nenhum vírus
existente tem esta sequência genética. Isto é estranho, porque para que um
vírus adquira uma grande sequência como esta, deve obtê-lo de outro organismo. Ele
não tem um laboratório para manipular sequências genéticas, nem os morcegos
(daí a piada de Jikky, o rato de laboratório)...

É muito fácil alterar
um único nucleótido (uma mutação pontual ou SNP) ou até mesmo inserir ou
remover nucleótidos (o que é menos comum) mas inserir 20 ou 30 nucleótidos com
um código que funciona? Não, deve ter vindo de outro vírus ou foi
feito em laboratório.
Então, de onde vem este código? Bem, acontece que blast pode-nos dizer –
com algum grau de certeza – de onde vem parte deste código, especialmente a
parte que codifica para a secção PRRAR (o local de clivagem de furina que é tão
único).
A parte que nos interessa está no índice Jikky. CTCCTCGGGGGGCACGTAG. Vamos
fazer uma BLAST

O que você vê são as
mesmas sequências (sars-CoV-2 mal classificado) nos primeiros 9 resultados, e
então nenhum dos resultados restantes mostra uma partida de 19/19. Isto
significa que
nenhum vírus conhecido pelos seres humanos tinha esta sequência particular no
seu genoma antes da descoberta de SARS-Cov-2. Então, de onde vem? Para isso, tem
de escolher outra base de dados. Voltemos ao ecrã de sondagens DA BLASTn e
alteremos a opção de base de dados para "Sequências de patentes (pat)". Retire todas as
exclusões e execute o BLAST.

Os resultados precisam de ser examinados, uma vez que o topo da lista
inclui os resultados das patentes deste ano. São pré-fixados WO2021 e WO2020 e
podem, portanto, ser ignorados. Logo abaixo estão as patentes que nos
interessam. Só realcei os três primeiros, mas toda a lista inclui muitas
patentes detidas pela mesma empresa. Basta clicar no número de adesão à
direita.

Então façamo-lo e vejamos que empresa, que todos nós sabemos (agora) ser uma empresa farmacêutica que nunca produziu um medicamento funcional e que no entanto tem uma capitalização de mercado de mais de 80 biliões de dólares...
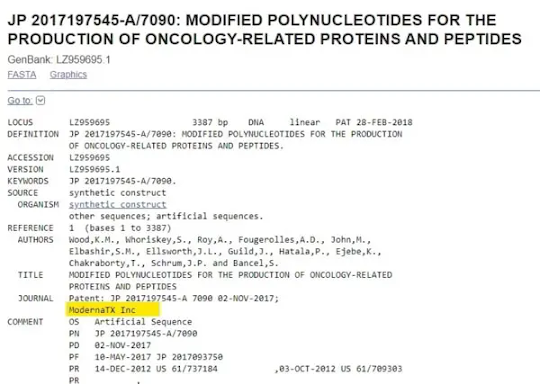
Sim, está certo. Cada uma destas patentes que contém esta sequência de 19nt (cuja probabilidade de aparecer por acaso é inferior a 1 em mil milhões) vem da Moderna. (Note-se que a sequência é na verdade a sequência complementar inversa, mas este é provavelmente um resultado directo das linhas celulares em que ocorreu – linhas celulares MSH3_transformadas (mutées, em francês – NdT) concebidas para desenvolver vacinas contra o cancro, a patente Moderna era na verdade sobre um gene MSH3 transformado para este fim.)
Para que esta sequência aparecesse neste
vírus, o vírus feito com as suas inserções de VIH tinha de ter sido infectado
em linhas celulares patenteadas fornecidas pela Moderna, que possuíam esta
sequência única não encontrada em nenhum outro vírus.
Em teoria, nada é impossível na ciência, na medicina ou na genómica. Um
vírus SARS que surge naturalmente com 3 inserções de HIV nos seus locais de ligação
e que também contém um local de clivagem de furina que não existe na natureza
mas que existe numa patente Moderna... É realmente qualquer coisa. Não existe.
Um elefante rosa voador seria um milhão de vezes mais provável.
§ Leia Também: Um especialista em genética ligado à OMS explica como o
Covid-19 poderia ter sido fabricado em laboratório
Este
artigo foi traduzido para Língua Portuguesa por Luis Júdice

Sem comentários:
Enviar um comentário